 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDirected Graph Convolutional Network
Papers and Code
The Effect of Architecture During Continual Learning
Jan 27, 2026Continual learning is a challenge for models with static architecture, as they fail to adapt to when data distributions evolve across tasks. We introduce a mathematical framework that jointly models architecture and weights in a Sobolev space, enabling a rigorous investigation into the role of neural network architecture in continual learning and its effect on the forgetting loss. We derive necessary conditions for the continual learning solution and prove that learning only model weights is insufficient to mitigate catastrophic forgetting under distribution shifts. Consequently, we prove that by learning the architecture and weights simultaneously at each task, we can reduce catastrophic forgetting. To learn weights and architecture simultaneously, we formulate continual learning as a bilevel optimization problem: the upper level selects an optimal architecture for a given task, while the lower level computes optimal weights via dynamic programming over all tasks. To solve the upper level problem, we introduce a derivative-free direct search algorithm to determine the optimal architecture. Once found, we must transfer knowledge from the current architecture to the optimal one. However, the optimal architecture will result in a weights parameter space different from the current architecture (i.e., dimensions of weights matrices will not match). To bridge the dimensionality gap, we develop a low-rank transfer mechanism to map knowledge across architectures of mismatched dimensions. Empirical studies across regression and classification problems, including feedforward, convolutional, and graph neural networks, demonstrate that learning the optimal architecture and weights simultaneously yields substantially improved performance (up to two orders of magnitude), reduced forgetting, and enhanced robustness to noise compared with static architecture approaches.
SAGE-FM: A lightweight and interpretable spatial transcriptomics foundation model
Jan 21, 2026Spatial transcriptomics enables spatial gene expression profiling, motivating computational models that capture spatially conditioned regulatory relationships. We introduce SAGE-FM, a lightweight spatial transcriptomics foundation model based on graph convolutional networks (GCNs) trained with a masked central spot prediction objective. Trained on 416 human Visium samples spanning 15 organs, SAGE-FM learns spatially coherent embeddings that robustly recover masked genes, with 91% of masked genes showing significant correlations (p < 0.05). The embeddings generated by SAGE-FM outperform MOFA and existing spatial transcriptomics methods in unsupervised clustering and preservation of biological heterogeneity. SAGE-FM generalizes to downstream tasks, enabling 81% accuracy in pathologist-defined spot annotation in oropharyngeal squamous cell carcinoma and improving glioblastoma subtype prediction relative to MOFA. In silico perturbation experiments further demonstrate that the model captures directional ligand-receptor and upstream-downstream regulatory effects consistent with ground truth. These results demonstrate that simple, parameter-efficient GCNs can serve as biologically interpretable and spatially aware foundation models for large-scale spatial transcriptomics.
Provably Convergent Decentralized Optimization over Directed Graphs under Generalized Smoothness
Jan 07, 2026Decentralized optimization has become a fundamental tool for large-scale learning systems; however, most existing methods rely on the classical Lipschitz smoothness assumption, which is often violated in problems with rapidly varying gradients. Motivated by this limitation, we study decentralized optimization under the generalized $(L_0, L_1)$-smoothness framework, in which the Hessian norm is allowed to grow linearly with the gradient norm, thereby accommodating rapidly varying gradients beyond classical Lipschitz smoothness. We integrate gradient-tracking techniques with gradient clipping and carefully design the clipping threshold to ensure accurate convergence over directed communication graphs under generalized smoothness. In contrast to existing distributed optimization results under generalized smoothness that require a bounded gradient dissimilarity assumption, our results remain valid even when the gradient dissimilarity is unbounded, making the proposed framework more applicable to realistic heterogeneous data environments. We validate our approach via numerical experiments on standard benchmark datasets, including LIBSVM and CIFAR-10, using regularized logistic regression and convolutional neural networks, demonstrating superior stability and faster convergence over existing methods.
Multi-fidelity graph-based neural networks architectures to learn Navier-Stokes solutions on non-parametrized 2D domains
Jan 05, 2026We propose a graph-based, multi-fidelity learning framework for the prediction of stationary Navier--Stokes solutions in non-parametrized two-dimensional geometries. The method is designed to guide the learning process through successive approximations, starting from reduced-order and full Stokes models, and progressively approaching the Navier--Stokes solution. To effectively capture both local and long-range dependencies in the velocity and pressure fields, we combine graph neural networks with Transformer and Mamba architectures. While Transformers achieve the highest accuracy, we show that Mamba can be successfully adapted to graph-structured data through an unsupervised node-ordering strategy. The Mamba approach significantly reduces computational cost while maintaining performance. Physical knowledge is embedded directly into the architecture through an encoding -- processing -- physics informed decoding pipeline. Derivatives are computed through algebraic operators constructed via the Weighted Least Squares method. The flexibility of these operators allows us not only to make the output obey the governing equations, but also to constrain selected hidden features to satisfy mass conservation. We introduce additional physical biases through an enriched graph convolution with the same differential operators describing the PDEs. Overall, we successfully guide the learning process by physical knowledge and fluid dynamics insights, leading to more regular and accurate predictions
Gene regulatory network inference algorithm based on spectral signed directed graph convolution
Dec 12, 2025Accurately reconstructing Gene Regulatory Networks (GRNs) is crucial for understanding gene functions and disease mechanisms. Single-cell RNA sequencing (scRNA-seq) technology provides vast data for computational GRN reconstruction. Since GRNs are ideally modeled as signed directed graphs to capture activation/inhibition relationships, the most intuitive and reasonable approach is to design feature extractors based on the topological structure of GRNs to extract structural features, then combine them with biological characteristics for research. However, traditional spectral graph convolution struggles with this representation. Thus, we propose MSGRNLink, a novel framework that explicitly models GRNs as signed directed graphs and employs magnetic signed Laplacian convolution. Experiments across simulated and real datasets demonstrate that MSGRNLink outperforms all baseline models in AUROC. Parameter sensitivity analysis and ablation studies confirmed its robustness and the importance of each module. In a bladder cancer case study, MSGRNLink predicted more known edges and edge signs than benchmark models, further validating its biological relevance.
The Affine Divergence: Aligning Activation Updates Beyond Normalisation
Dec 24, 2025A systematic mismatch exists between mathematically ideal and effective activation updates during gradient descent. As intended, parameters update in their direction of steepest descent. However, activations are argued to constitute a more directly impactful quantity to prioritise in optimisation, as they are closer to the loss in the computational graph and carry sample-dependent information through the network. Yet their propagated updates do not take the optimal steepest-descent step. These quantities exhibit non-ideal sample-wise scaling across affine, convolutional, and attention layers. Solutions to correct for this are trivial and, entirely incidentally, derive normalisation from first principles despite motivational independence. Consequently, such considerations offer a fresh and conceptual reframe of normalisation's action, with auxiliary experiments bolstering this mechanistically. Moreover, this analysis makes clear a second possibility: a solution that is functionally distinct from modern normalisations, without scale-invariance, yet remains empirically successful, outperforming conventional normalisers across several tests. This is presented as an alternative to the affine map. This generalises to convolution via a new functional form, "PatchNorm", a compositionally inseparable normaliser. Together, these provide an alternative mechanistic framework that adds to, and counters some of, the discussion of normalisation. Further, it is argued that normalisers are better decomposed into activation-function-like maps with parameterised scaling, thereby aiding the prioritisation of representations during optimisation. Overall, this constitutes a theoretical-principled approach that yields several new functions that are empirically validated and raises questions about the affine + nonlinear approach to model creation.
Insider Threat Detection Using GCN and Bi-LSTM with Explicit and Implicit Graph Representations
Dec 20, 2025



Insider threat detection (ITD) is challenging due to the subtle and concealed nature of malicious activities performed by trusted users. This paper proposes a post-hoc ITD framework that integrates explicit and implicit graph representations with temporal modelling to capture complex user behaviour patterns. An explicit graph is constructed using predefined organisational rules to model direct relationships among user activities. To mitigate noise and limitations in this hand-crafted structure, an implicit graph is learned from feature similarities using the Gumbel-Softmax trick, enabling the discovery of latent behavioural relationships. Separate Graph Convolutional Networks (GCNs) process the explicit and implicit graphs to generate node embeddings, which are concatenated and refined through an attention mechanism to emphasise threat-relevant features. The refined representations are then passed to a bidirectional Long Short-Term Memory (Bi-LSTM) network to capture temporal dependencies in user behaviour. Activities are flagged as anomalous when their probability scores fall below a predefined threshold. Extensive experiments on CERT r5.2 and r6.2 datasets demonstrate that the proposed framework outperforms state-of-the-art methods. On r5.2, the model achieves an AUC of 98.62, a detection rate of 100%, and a false positive rate of 0.05. On the more challenging r6.2 dataset, it attains an AUC of 88.48, a detection rate of 80.15%, and a false positive rate of 0.15, highlighting the effectiveness of combining graph-based and temporal representations for robust ITD.
GFLAN: Generative Functional Layouts
Dec 18, 2025Automated floor plan generation lies at the intersection of combinatorial search, geometric constraint satisfaction, and functional design requirements -- a confluence that has historically resisted a unified computational treatment. While recent deep learning approaches have improved the state of the art, they often struggle to capture architectural reasoning: the precedence of topological relationships over geometric instantiation, the propagation of functional constraints through adjacency networks, and the emergence of circulation patterns from local connectivity decisions. To address these fundamental challenges, this paper introduces GFLAN, a generative framework that restructures floor plan synthesis through explicit factorization into topological planning and geometric realization. Given a single exterior boundary and a front-door location, our approach departs from direct pixel-to-pixel or wall-tracing generation in favor of a principled two-stage decomposition. Stage A employs a specialized convolutional architecture with dual encoders -- separating invariant spatial context from evolving layout state -- to sequentially allocate room centroids within the building envelope via discrete probability maps over feasible placements. Stage B constructs a heterogeneous graph linking room nodes to boundary vertices, then applies a Transformer-augmented graph neural network (GNN) that jointly regresses room boundaries.
CheXmask-U: Quantifying uncertainty in landmark-based anatomical segmentation for X-ray images
Dec 11, 2025Uncertainty estimation is essential for the safe clinical deployment of medical image segmentation systems, enabling the identification of unreliable predictions and supporting human oversight. While prior work has largely focused on pixel-level uncertainty, landmark-based segmentation offers inherent topological guarantees yet remains underexplored from an uncertainty perspective. In this work, we study uncertainty estimation for anatomical landmark-based segmentation on chest X-rays. Inspired by hybrid neural network architectures that combine standard image convolutional encoders with graph-based generative decoders, and leveraging their variational latent space, we derive two complementary measures: (i) latent uncertainty, captured directly from the learned distribution parameters, and (ii) predictive uncertainty, obtained by generating multiple stochastic output predictions from latent samples. Through controlled corruption experiments we show that both uncertainty measures increase with perturbation severity, reflecting both global and local degradation. We demonstrate that these uncertainty signals can identify unreliable predictions by comparing with manual ground-truth, and support out-of-distribution detection on the CheXmask dataset. More importantly, we release CheXmask-U (huggingface.co/datasets/mcosarinsky/CheXmask-U), a large scale dataset of 657,566 chest X-ray landmark segmentations with per-node uncertainty estimates, enabling researchers to account for spatial variations in segmentation quality when using these anatomical masks. Our findings establish uncertainty estimation as a promising direction to enhance robustness and safe deployment of landmark-based anatomical segmentation methods in chest X-ray. A fully working interactive demo of the method is available at huggingface.co/spaces/matiasky/CheXmask-U and the source code at github.com/mcosarinsky/CheXmask-U.
Graph-Based Learning of Spectro-Topographical EEG Representations with Gradient Alignment for Brain-Computer Interfaces
Dec 08, 2025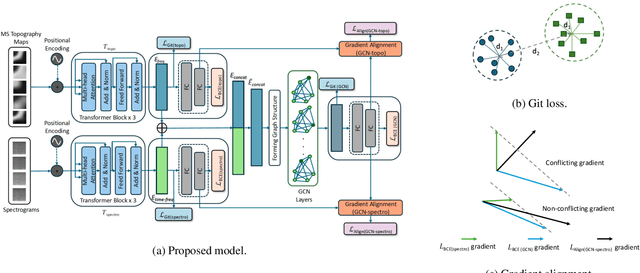
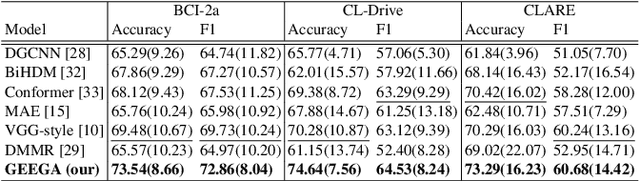
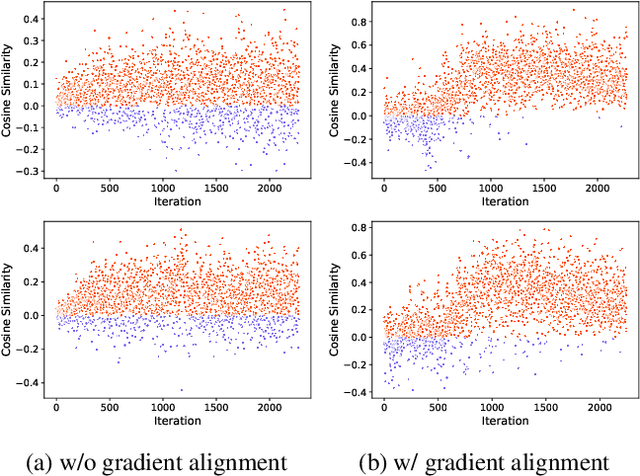
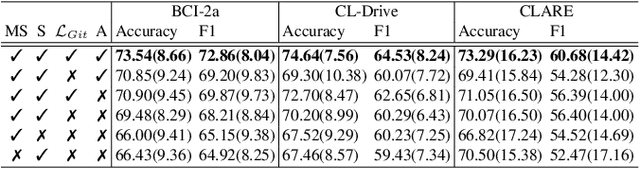
We present a novel graph-based learning of EEG representations with gradient alignment (GEEGA) that leverages multi-domain information to learn EEG representations for brain-computer interfaces. Our model leverages graph convolutional networks to fuse embeddings from frequency-based topographical maps and time-frequency spectrograms, capturing inter-domain relationships. GEEGA addresses the challenge of achieving high inter-class separability, which arises from the temporally dynamic and subject-sensitive nature of EEG signals by incorporating the center loss and pairwise difference loss. Additionally, GEEGA incorporates a gradient alignment strategy to resolve conflicts between gradients from different domains and the fused embeddings, ensuring that discrepancies, where gradients point in conflicting directions, are aligned toward a unified optimization direction. We validate the efficacy of our method through extensive experiments on three publicly available EEG datasets: BCI-2a, CL-Drive and CLARE. Comprehensive ablation studies further highlight the impact of various components of our model.